

Visual Abstract

Paroxysmal nocturnal hemoglobinuria (PNH) and primary complement–mediated thrombotic microangiopathy, also known as atypical hemolytic uremic syndrome (aHUS), are hematologic disorders characterized by dysregulation of the complement system leading to hemolysis and other systemic and potentially lethal complications. The advent of C5 inhibition with agents such as eculizumab and ravulizumab has revolutionized the management of patients with these disorders, although some may still experience clinically significant breakthrough extravascular hemolysis. Over the past several years, novel therapies targeting upstream pathways of complement inhibition have been approved for the treatment of patients with PNH experiencing breakthrough hemolysis despite C5 inhibition. These agents currently include pegcetacoplan, a C3 inhibitor; iptacopan, an oral factor B inhibitor; danicopan, an oral factor D inhibitor; and crovalimab, an anti-C5 monoclonal antibody. This review highlights recent advances in complement-targeted therapies for PNH, including their indications and efficacy and safety data from key randomized trials. In addition, we review current data supporting the use of these novel agents for aHUS, for which only the terminal complement inhibitors eculizumab and ravulizumab are currently approved. Future research is crucial to establish the long-term efficacy and safety profiles of these novel therapies, ensuring the best treatment strategies for patients with PNH and aHUS.
Introduction
Paroxysmal nocturnal hemoglobinuria (PNH) is a rare, acquired clonal hematopoietic disorder caused by a deficiency in the complement inhibitory proteins CD55 and CD59.1 The absence of these proteins on hematopoietic stem cells makes them vulnerable to complement-mediated destruction, resulting in intravascular and extravascular hemolysis, potentially leading to life-threatening complications.1-3
Historically, the first-line treatment for PNH targets complement-mediated intravascular hemolysis using C5 inhibitors, eculizumab and ravulizumab, blocking the late stages of the complement cascade and preventing membrane attack complex formation.4 However, it is well documented that some patients with PNH continue to experience breakthrough hemolysis (BTH), attributable to dysregulation of the alternative pathway.5 This disturbance leads to accumulation of C3 opsonins on PNH erythrocytes, that are then recognized by complement receptors on macrophages, resulting in phagocytosis and subsequent extravascular hemolysis.5 As a result, new complement inhibitors have been developed to target upstream pathways of the complement system, demonstrating significant potential in tackling these challenges. It is worth noting that complement inhibition increases susceptibility to Neisseria meningitidis and other encapsulated bacteria. Therefore, patients should be vaccinated or receive prophylactic antibiotics if vaccination is delayed.3,6
Complement-mediated thrombotic microangiopathy (CM-TMA), triggered by classical pathway activation and amplified through the alternative pathway, leads to hemolytic anemia, thrombocytopenia, and organ damage.7,8 Primary CM-TMA (also known as atypical hemolytic uremic syndrome [aHUS]) results from genetic complement variants, whereas secondary CM-TMA arises from triggers such as infection, malignancy, hematopoietic stem cell transplantation (HSCT), drugs, autoimmune disorders, or pregnancy.5,9 Approximately half of patients with primary CM-TMA lack identifiable mutations, complicating diagnosis and treatment.5,9,10 Despite significant improvements in aHUS outcomes with terminal C5 inhibition, many patients remain transfusion dependent.3 The development of novel alternative complement inhibitors may enhance aHUS treatment outcomes, although current data supporting their routine use remain limited.
This review summarizes the latest complement inhibitor therapies and clinical trial data to inform clinicians on treatment options for PNH and aHUS. It contrasts terminal complement inhibitors (eculizumab, ravulizumab, and crovalimab) with alternative pathway inhibitors (pegcetacoplan, iptacopan, and danicopan) for PNH and discusses complement inhibitors in aHUS, including narsoplimab for HSCT-associated TMA. Management of secondary TMA syndromes is beyond the scope of this review and will not be fully addressed.
Methods
A systematic literature search was conducted using PubMed to identify studies evaluating the use of anticomplement medications in patients with PNH experiencing BTH and in those diagnosed with primary CM-TMA. The search strategy included a combination of medical subject headings terms and relevant keywords, such as “paroxysmal nocturnal hemoglobinuria,” “breakthrough hemolysis,” “complement-mediated thrombotic microangiopathy,” and “anti-complement therapy.” Studies were included if they specifically assessed the efficacy, safety, or clinical outcomes of anticomplement therapies in these conditions. Case reports, case series, retrospective and prospective cohort studies, and clinical trials were all considered for inclusion. Studies were excluded if they focused on patients without documented BTH in PNH or if they primarily investigated secondary CM-TMA, because these conditions fall outside the scope of this review. Articles that did not provide specific clinical data related to anticomplement therapy were also excluded.
Results
Eculizumab
Eculizumab, a C5 inhibitor, was the first approved complement-directed therapy for patients with PNH and aHUS and has been associated with reduced morbidity and mortality in this patient population11,12 (Table 1). Eculizumab is administered IV, with a loading dose of 600 mg weekly for 4 doses, followed by a maintenance dose of 900 mg every 2 weeks11 (Table 2). Eculizumab is a monoclonal antibody that targets the C5 protein, blocking its activation and preventing the formation of the terminal attack complex12,13 (Figure 1). Although eculizumab is well tolerated, it carries a 0.5% annual risk of contracting N meningitidis infection.13,14 Leading studies include the TRIUMPH (N = 87) and SHEPHERD (N = 97) trials, reporting hemoglobin stabilization, reduced red blood cell transfusions, decreased hemolysis, and improved fatigue, among patients with PNH7,15 (Table 2). Importantly, eculizumab is the only approved therapy to treat women with PNH during pregnancy, reducing mortality and the incidence of thrombotic events.15,16
Summary of end points of key trials of novel anticomplement-directed therapies in patients with PNH
| Anticomplement-directed therapy | Mean increase in Hb (g/dL) | Percent of participants achieving Hb stabilization, % | Decrease in LDH (U/L) | Transfusion avoidance, % | BTH, % | Mean change in FACIT-Fatigue scores |
|---|---|---|---|---|---|---|
| Iptacopan17-20 | 2.0-3.59 | — | ≤1.5 × ULN | 95.2 | — | 8.6-10.8 |
| Pegcetacoplan7,8,21-24 | 2.4-3.84 | 85 | ≤1.5 × ULN | 85-91 | 10-19 | 11-32 |
| Danicopan25,26 | 2.4-2.94 | — | 1.8 × ULN | 83-91 | — | 8.0-11 |
| Crovalimab27-29 | 1.0-1.2 | 56-63 | ≤1.5 × ULN | 65-82 | 4.0-10.4 | 7.8 |
| Eculizumab7,11,15 | 0.8 | 49-60 | 1.25 × ULN | 68-82 | 5.1-23 | 5.2-12 |
| Ravulizumab4,30 | — | 68-78 | ≤1.5 × ULN | 73-86 | 0-4 | 6-10 |
| Anticomplement-directed therapy | Mean increase in Hb (g/dL) | Percent of participants achieving Hb stabilization, % | Decrease in LDH (U/L) | Transfusion avoidance, % | BTH, % | Mean change in FACIT-Fatigue scores |
|---|---|---|---|---|---|---|
| Iptacopan17-20 | 2.0-3.59 | — | ≤1.5 × ULN | 95.2 | — | 8.6-10.8 |
| Pegcetacoplan7,8,21-24 | 2.4-3.84 | 85 | ≤1.5 × ULN | 85-91 | 10-19 | 11-32 |
| Danicopan25,26 | 2.4-2.94 | — | 1.8 × ULN | 83-91 | — | 8.0-11 |
| Crovalimab27-29 | 1.0-1.2 | 56-63 | ≤1.5 × ULN | 65-82 | 4.0-10.4 | 7.8 |
| Eculizumab7,11,15 | 0.8 | 49-60 | 1.25 × ULN | 68-82 | 5.1-23 | 5.2-12 |
| Ravulizumab4,30 | — | 68-78 | ≤1.5 × ULN | 73-86 | 0-4 | 6-10 |
Hb, hemoglobin.
Drug formulation and dosing reference
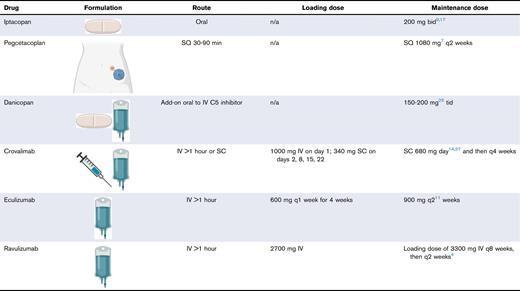
bid, twice daily; n/a, not available; q, every; SQ, SC infusion pump; tid, 3 times per day.

Mechanism of action of complement inhibitors and their effects on the complement cascade. FB, factor B; FD, factor D; MAC, complement membrane attack complex.
Mechanism of action of complement inhibitors and their effects on the complement cascade. FB, factor B; FD, factor D; MAC, complement membrane attack complex.
Ravulizumab
Ravulizumab was designed to address limitations in eculizumab therapy, particularly eculizumab’s short half-life and frequent dosing schedule.30 Ravulizumab, a long-acting C5 inhibitor with 4 times longer mean half-life than eculizumab, provides immediate and sustained C5 inhibition over 8-week dosing intervals.30 The 301 and 302 trials are 2 key studies that evaluated the efficacy and safety of ravulizumab for the treatment of PNH.4,30,31 The 301 study randomized 246 complement-naïve participants to receive either ravulizumab or eculizumab, with a 26-week follow-up.4,13,15,31 The results showed that ravulizumab was associated with a lower incidence of BTH and a higher rate of transfusion avoidance, with 81% of participants in the ravulizumab group avoiding transfusions.4,15 In the 302 study, 195 participants who had previously achieved stability on eculizumab were randomized into 2 groups, to continue eculizumab vs switching to ravulizumab.15,30,31 Eighty-five percent of participants receiving ravulizumab achieved higher rates of transfusion independence, with no patients in the ravulizumab group experiencing BTH compared with 5 patients (5.1%) in the eculizumab group.15,30,31
At this time, the anti-C5–directed therapies remain the only 2 medications approved for the treatment of aHUS.32 Both eculizumab and ravulizumab have shown significant clinical benefits in resolving markers of TMA in aHUS, as measured by improvements in kidney function and normalization of platelet count and lactate dehydrogenase (LDH).32,33 At 26 weeks, 56% of adults on eculizumab and 54% on ravulizumab achieved a complete TMA response, with no significant differences in outcomes or safety between treatments33 Despite these efforts, a small percentage of patients remain transfusion dependent, because C5 inhibitors target intravascular but not C3-mediated extravascular hemolysis, resulting in incomplete response and BTH.8,30 Currently, there is no published, high-quality data supporting the use of novel alternative pathway inhibitors for treatment of aHUS, however ongoing studies hold promise for future advancements.
Crovalimab
Crovalimab, a novel monoclonal antibody that binds to an epitope on the C5 β-chain, in contrast to eculizumab and ravulizumab, that target the C5 α-chain.34 Crovalimab demonstrates noninferiority to other anti-C5 therapies and provides the convenience of monthly subcutaneous (SC) self-administration after an initial IV infusion (Table 1).34 In the COMMODORE 1 trial, 84 participants who had been on eculizumab for ≥6 months were randomized to either continue eculizumab or switch to crovalimab for 24 weeks; crovalimab was shown to be noninferior to eculizumab in controlling hemolysis, avoiding transfusions, and preventing breakthrough hemolysis for 24 weeks.27,35 Crovalimab demonstrated efficacy comparable with that of eculizumab in controlling hemolysis (79.3% vs 79.0%), transfusion avoidance (65% vs 68%), BTH (10.4% vs 14.5%), and hemoglobin stabilization (63% vs 60.9%), offering an effective alternative with more convenient dosing.27,28,35 Both treatments had similar safety profiles, with most reactions being mild to moderate infusion-related events, and no meningococcal infections or deaths were reported.28,35 Interestingly, most participants who switched from eculizumab to crovalimab preferred crovalimab because of reduced clinic visits, shorter transfusion times, and improved fatigue scores.27,35 Lastly, the phase 3 COMMODORE 3 trial (N = 50) assessed complement inhibitor–naïve patients with PNH, characterized by LDH >2× upper limit of normal (ULN) and ≥4 transfusions in 12 months; crovalimab achieved hemolysis control in 78% of participants, reducing LDH to <1.5× ULN.36 Transfusion avoidance was achieved in half of the participants, and those who required transfusions saw an average reduction of 6.1 units of packed red blood cells over the 24-week period.36 The COMPOSER study was a 3-part, open-label phase 1/2 trial that evaluated the safety, pharmacokinetics, and efficacy of crovalimab compared with eculizumab as well as the effect of switching from eculizumab to crovalimab.29 The key findings of COMPOSER 1, 2 and 3 revealed that 79% of the participants experienced a reduction in LDH levels, and 65% of those treated with crovalimab avoided transfusions compared with 68% in the eculizumab group.29 Crovalimab showed lower rates of BTH than those receiving eculizumab (10.4% vs 14.5%).29 Overall, both treatments had similar safety profiles, with marginally higher rates of nephrolithiasis and cholecystitis, that did not lead to discontinuation of this drug.29 These studies confirm crovalimab’s noninferiority to eculizumab, with less frequent infusions, but its role in the current treatment landscape remains unclear because of the lack of head-to-head comparisons with ravulizumab.
Pegcetacoplan
Pegcetacoplan is a pegylated peptide inhibitor that targets the proximal C3 protein, preventing C3 convertase formation and downstream complement activation, and is administered SC twice weekly via an infusion pump (Figure 1; Table 2).7,21,37 This mechanism allows pegcetacoplan to control both complement-mediated intravascular hemolysis and C3b-mediated extravascular hemolysis, which is of value in patients experiencing breakthrough extravascular hemolysis despite C5 inhibitor therapy.21,37 The literature defines clinical BTH as a decline in hemoglobin of ≥2 g/dL or development of clinical signs or symptoms of hemolysis, in combination with an increase in LDH of >1.5× ULN15,17,38 Potential triggers of BTH despite complement inhibitor therapy have been associated with recent infections or vaccinations, leading to activation of the complement pathway.22
The PADDOCK and PALOMINO trials were conducted in parallel to assess the safety and preliminary efficacy of pegcetacoplan in patients with PNH. The PADDOCK study was a phase 1b, multiple-ascending-dose pilot trial with 2 cohorts, whereas PALOMINO was a phase 2a single-cohort trial using the same protocol and dosing structure as the PADDOCK cohort 2 trial allowing for comparative results.22,23 The PADDOCK trial enrolled 3 patients in cohort 1 who received 180 mg/d of pegcetacoplan and 20 patients in cohort 2 who received 360 mg/d. The PALOMINO trial enrolled 4 patients, all receiving 360 mg/d of pegcetacoplan.23 After 1 year of starting pegcetacoplan, 65% of PADDOCK participants and all participants in the PALOMINO trial achieved transfusion independence.22,23 Initially, both studies reported hemoglobin levels below normal, but by 1 year of treatment, the mean increase in hemoglobin was 3 g/dL in comparison with the control group.23 Although the PADDOCK trial reported 13 serious adverse events (SAEs), including aplastic anemia, abdominal neoplasm, and hypersensitivity, leading to discontinuation in 3 patients, pegcetacoplan demonstrated sufficient efficacy and safety, supporting further phase 3 trials.23
The PRINCE trial was a phase 3 randomized, multicenter study designed to evaluate the efficacy of pegcetacoplan compared with placebo in complement inhibitor–naïve patients with PNH. Trial results demonstrated that among patients receiving pegcetacoplan (n = 35), 85.7% demonstrated hemoglobin stabilization by 16 weeks compared with 0% in the control group (n = 18; P < .0001).24,39 In addition, LDH levels were significantly reduced by week 4 and remained stable through week 26.24,39 Pegcetacoplan also showed superiority in transfusion independence, with 91.4% of patients avoiding transfusions, compared with just 5.6% in the control group, with a significantly lower number of transfused packed red blood cell units.24,39 Despite these significant results, the study has not yet reported safety and side effect data.
The PEGASUS trial (N = 80) was another open-label, phase 3 randomized controlled study comparing pegcetacoplan as a standalone therapy vs eculizumab after an initial 4-week run-in phase of both eculizumab and pegcetacoplan for patients with ongoing anemia despite eculizumab therapy.7 By week 16, those receiving pegcetacoplan had significantly increased hemoglobin levels by a mean of 3.84 g/dL from baseline (P < .001).7,14 In addition, 85% of patients on pegcetacoplan achieved transfusion independence, compared with just 15% in the eculizumab group.7 Pegcetacoplan was associated with a similar reduction in hemolysis markers compared with eculizumab, although with a lower rate of BTH (10% vs 23%).7 In addition, sustained responses, including improvement in hemoglobin, fatigue reduction, and reduced need for transfusions, were achieved over 48 weeks.7 The most common AEs for all patients receiving pegcetacoplan monotherapy during the open-label period were injection site reactions, diarrhea, headache, nasopharyngitis, fatigue, and BTH.7
BTH is of noteworthy concern in patients with PNH who receive pegcetacoplan because of the potential for hemolytic events of greater severity with proximal complement inhibition, as well as generally less guidance on the optimal management.21 Pegcetacoplan’s C3 inhibition reduces hemolysis and prolongs PNH red blood cell survival but may also increase susceptibility to lysis, potentially causing severe anemia.7 If severe anemia occurs, the PEGASUS trial suggests immediate transfusion, pegcetacoplan dose adjustment, or addition of eculizumab to control hemolysis and prevent complications, while continuing pegcetacoplan therapy.7 The OLE study, a multicenter trial of patients with PNH from 5 previous pegcetacoplan studies, explores managing BTH, including acute IV or intensive SC dosing. Per protocol, pegcetacoplan dosing can increase from twice to 3 times weekly if LDH exceeds twice the ULN.22 Patients with BTH were eligible for intensive treatment, either a single 1080 mg IV dose or 1080 mg SC for 3 days.22 This approach rapidly reduced LDH, stabilized hemoglobin, with full resolution of BTH in all patients.22 Although these findings are promising for improving management of BTH in PNH with proximal C3 complement inhibition, there are currently no data supporting the use of pegcetacoplan for the treatment of aHUS, and we await future research to explore its potential therapeutic use.
Iptacopan
Iptacopan is an oral serine protease inhibitor targeting factor B in the alternative complement pathway, blocking the activation of the C3 convertase enzyme. This inhibition disrupts C3-dependent opsonization, anaphylatoxin-induced inflammatory cell recruitment, and the formation of the membrane attack complex, therefore preventing both intravascular and extravascular hemolysis and potentially offering a therapeutic advantage over traditional C5 inhibitors18,37 (Figures 1 and 2).

A recent phase 2 study investigated iptacopan monotherapy in 12 treatment-naïve patients with active hemolysis secondary to PNH, reporting statistically significant improvements in hemoglobin and a reduction in hemolysis markers after 12 weeks.17,19 In a separate multicenter, open-label, phase 2 trial, 10 patients with residual hemolysis despite eculizumab therapy received iptacopan as add-on therapy.17,19 This study demonstrated a mean hemoglobin increase of 3.19 g/dL and a mean reduction in LDH of −309.2 IU/L after 13 weeks, with no SAEs.17 After 6 months, 7 of 10 participants discontinued eculizumab without evidence of BTH or hemoglobin decline.17 Both studies highlight iptacopan’s efficacy in normalizing hemoglobin and reducing LDH, offering a safe and convenient oral option for C3–mediated extravascular and membrane attack complex–driven hemolysis. However, limitations include the lack of control groups and baseline variability
Most importantly, 2 key phase 3 trials assessed the efficacy and safety of iptacopan as monotherapy for treatment-naïve patients with PNH. The APPOINT-PNH trial found that most participants (31/33) experienced a hemoglobin increase of 2 g/dL from baseline within the 24-week study period.40 None of the 33 participants required a red cell transfusion, reported BTH, or experienced any SAEs.40 Notably, LDH levels decreased by an average of 83%, reticulocyte counts dropped by 82%, and bilirubin levels decreased by 18.1 μmol/L.40 Importantly, participants experienced an average increase of 10.8 points on the FACIT-Fatigue (Functional Assessment of Chronic Illness Therapy–Fatigue) scale (range, 0-52), highlighting positive impact on quality of life.40 The most common AEs of iptacopan included headache and increased risk of contracting COVID-19 and other upper respiratory tract infections.40
The second phase 3 trial of iptacopan, the APPLY-PNH trial, randomized participants who remained anemic despite receiving at least 6 months of either eculizumab or ravulizumab to either iptacopan (n = 62) or to continue anti-C5–directed therapy (n = 35) for 24 weeks.40 Among patients treated with iptacopan, 85% experienced a hemoglobin increase of at least 2 g/dL compared with no patients on C5-inhibitor therapy.40 Of 62 patients who received iptacopan, 2 experienced clinical BTH events, whereas 6 of 35 patients who received anti-C5 therapy had similar occurrences.40 Iptacopan demonstrated superior efficacy in reducing the need for blood transfusions, with 95% of participants receiving iptacopan avoiding transfusions, compared with only 40% in the group receiving anti-C5 therapy.40 The secondary end points to this study reported no significant difference in the percentage change of LDH, however there was a statistically significant mean reduction in absolute reticulocyte count among those on iptacopan.40 Lastly, participants on iptacopan reported an average improvement of 8.6 points in the FACIT-Fatigue score, whereas those on anti-C5 therapy showed almost no change (0.3 points).17 There was also a lower rate of SAEs among those on iptacopan vs anti-C5 therapy, 10% compared with 14%, respectively.40 The trial results suggest that iptacopan improves hemoglobin levels, reduces transfusion dependence, and improves fatigue more effectively than anti-C5 inhibitors.
Notable limitations of iptacopan include its short half-life requiring twice daily dosing. Limited diversity in APPLY-PNH, including low Black patient recruitment, may affect generalizability given racial differences in thrombosis risk. Longer follow-up is needed to better assess infection rates and BTH, as the optimal management of BTH with iptacopan is yet to be determined. Data from the 48-week APPLY-PNH and APPOINT-PNH trials indicate that 9 of 136 patients on iptacopan experienced BTH, a significantly lower incidence compared with those treated with anti-C5 therapies.40 Of 9 events of clinical BTH, all were mild to moderate, except 1 severe case associated with COVID-19 and the development of cold agglutinins.40 No patient discontinued iptacopan, and red blood cell transfusion was the most common intervention, with only 1 patient receiving eculizumab. Cumulatively, these findings suggest that BTH on iptacopan was manageable and did not necessitate treatment discontinuation or intensification, in contrast to the more severe BTH observed with C5 inhibitors or pegcetacoplan.
Although iptacopan has not yet received US Food and Drug Administration approval for aHUS, it is currently being investigated in several active clinical trials. The APPELHUS trial (ClinicalTrials.gov identifier: NCT04889430), an ongoing multicenter, single-arm study enrolling complement inhibitor–naïve patients, aims to evaluate the efficacy and safety of iptacopan in adults with aHUS.20 Additional trials, including an open-label extension study (ClinicalTrials.gov identifier: NCT05795140), as well as a trial investigating the safety and efficacy of switching from anti-C5 therapy to iptacopan (ClinicalTrials.gov identifier: NCT05935215), are actively being recruited. If successful, iptacopan and similar alternative pathway inhibitors might improve outcomes and change the treatment landscape for aHUS in the near future.
Danicopan
Danicopan is an oral medication designed to target and inhibit factor D, a key enzyme in the alternative complement pathway (Figure 1), reducing excessive hemolysis in patients with PNH.25 Danicopan and iptacopan both inhibit C3 convertase formation, but danicopan acts by targeting factor D, whereas iptacopan inhibits factor B. Although both are administered orally, danicopan’s shorter half-life necessitates thrice-daily dosing, which may pose a limitation. Unlike iptacopan, which can be used as monotherapy, danicopan is specifically designed as an adjunct to C5 inhibitors, such as ravulizumab or eculizumab, for patients with PNH experiencing clinically significant BTH.26 Promising evidence in the literature suggests that this combination increases mean hemoglobin levels, reduces the frequency of BTH, and lowers the need for blood transfusions in patients with PNH.25,26
Initial data to support the use of danicopan comes from a phase 2 dose-finding trial involving 12 transfusion-dependent patients with PNH. Combination therapy of eculizumab and danicopan was associated with a mean increase in hemoglobin levels of 2.4 g/dL (P = .0001), and transfusion requirements were significantly reduced, with only 1 transfusion administered to 1 of 12 participants.25,41 Secondary findings included a decrease in the mean reticulocyte count and total bilirubin levels, as well as an increase of 11 points in the mean FACIT-Fatigue score25 (Table 1). Danicopan was generally well tolerated, with low-grade side effects such as headache, cough, nasopharyngitis, arthralgia, and fatigue, and no SAEs led to study withdrawal.25 Limitations included its single-center design, which limits external validity, a small sample size, and the absence of a control group, making it difficult to attribute the observed benefits solely to danicopan.
The ALPHA trial is a phase 3, double-blind study comparing danicopan plus eculizumab or ravulizumab with anti-C5 therapy alone in patients with persistent extravascular hemolysis despite 6 months of eculizumab or ravulizumab.41 The addition of danicopan plus either eculizumab or ravulizumab was associated with significantly increased hemoglobin levels as early as week 1, with an average rise of 2.94 g/dL compared with just 0.50 g/dL with C5 inhibitor monotherapy (P < .0001).41 Transfusion avoidance through week 12 was achieved in 83% of participants on combination danicopan plus C5 inhibition vs 38% for anti-C5 therapy alone.41 The FACIT-Fatigue score increased by 8.0 points with add-on danicopan, compared with 1.9 points with anti-C5 and placebo.41 The combination therapy was well tolerated, with headache, transaminitis, and cholecystitis as the most common adverse effects.26 Two participants discontinued owing to severe transaminitis; and no deaths, meningococcal infections, or discontinuations because of BTH were observed.26 Overall, danicopan, when used with C5 inhibitor therapy, improves hemoglobin levels and reduces transfusion requirements, although caution is warranted in patients with baseline liver disease.41 Long-term follow-up is necessary to assess sustained efficacy, safety, and appropriate patient selection. Furthermore, danicopan is not currently approved for aHUS, necessitating additional clinical studies to evaluate its safety and efficacy in this population.
Narsoplimab
Narsoplimab, a novel mannan-binding lectin serine protease 2 (MASP-2) inhibitor of the lectin pathway, shows potential therapeutic benefit in HSCT-associated TMA (HSCT-TMA).42 Post-HSCT endothelial injury, primarily because of increased risks from immunosuppression and infection, can directly activate the lectin pathway, with MASP-2, an effector enzyme, being elevated in patients with HSCT-TMA.42 A recent single-arm, open-label study of narsoplimab focused on primary HSCT-TMA, with 17 of 28 patients enrolled demonstrating response to narsoplimab on the basis of improved laboratory data and clinical benefit correlating with an estimated response rate of 61%.42 Transfusion independence was achieved in 48% of the population. The most reported AEs included infection in 71% of patients, as well as pyrexia, gastrointestinal side effects, including diarrhea, nausea, vomiting, and hypokalemia.42 Narsoplimab is not yet approved for the treatment for primary CM-TMA; further randomized control trials, including long-term follow-up data, will be required to understand the efficacy and safety in the HSCT population. We suspect that narsoplimab may play a role in other CM-TMA syndromes, especially those driven by direct endothelial injury, and eagerly await future clinical trials for these indications.
Discussion
Persistent anemia due to C3-mediated extravascular hemolysis remains a risk among patients with PNH treated with C5 inhibitor therapy, with only 20% to 30% of individuals achieving normal or near-normal hemoglobin levels.43 Interestingly, this variability in treatment response may be due to genetic differences, with studies suggesting that individuals with molecular variations may have a weaker response to C5-directed inhibition.43 To address these challenges, novel complement inhibitors have been developed to address BTH, showing early promise in therapeutic efficacy. The pivotal PEGASUS trial of pegcetacoplan, demonstrated a modest improvement in improving hemoglobin and reduced transfusion dependence compared with eculizumab, although with concern for potentially greater BTH and risk for infection.7,10 Preliminary data from the OLE study of pegcetacoplan suggest that BTH can be severe and may be best managed with an intensive administration protocol of pegcetacoplan, sometimes in addition to red blood cell transfusion and eculizumab therapy.22 Conversely, data from the APPLY-PNH and APPOINT-PNH studies suggest that iptacopan is superior to traditional anti-C5 inhibitor therapy in achieving significant increases in hemoglobin and resolution of extravascular hemolysis, with the added benefit of oral administration.8,40 BTH with iptacopan is infrequent and typically mild to moderate, most often managed with red blood cell transfusion without requiring drug discontinuation.8,40 Studies of danicopan, an oral factor D inhibitor, has reported improved hemoglobin and reduced transfusion requirements when used in addition to eculizumab or ravulizumab, although caution must be taken in those with baseline liver disease.25 Lastly, crovalimab a novel C5 inhibitor, provides a similar mechanism to eculizumab but with the flexibility of SC administration every 4 weeks compared with every 2 or 8 week IV infusions with eculizumab or ravulizumab, respectively.44 Additional therapeutic targets, including narsoplimab, the MASP-2 inhibitor of the lectin pathway, exemplify the rapidly evolving field of complement inhibitor therapy spanning across secondary causes of CM-TMA, such as HSCT-TMA, as well as other diseases driven by complement activation.5 Another significant consideration driving therapeutic selection is the substantially high cost of these agents, creating significant barriers to care. Eculizumab, ravulizumab, pegcetacoplan, and iptacopan have similar annual costs, ranging from $474 284 to $569 140 for eculizumab and ravulizumab, $516 880 to $556 932 for pegcetacoplan,45-47 and approximately $550 377 for iptacopan.48 Although the official price of danicopan has not been disclosed, a Canadian study estimates its annual cost to be between $75 000 and $100 000.46 Crovalimab’s wholesale price is reported at $21 228 per 2-mL vial.49 It remains unclear in this evolving treatment landscape whether the prohibitively high price of these agents will continue to be a major limitation, or if additional approved options may 1 day drive down costs. These novel drugs offer promising alternatives for patients with inadequate responses to C5 inhibitors. However, with limited long-term data, shared decision-making based on cost, administration, and efficacy is crucial for selecting complement therapies. Although emerging therapies show promise in managing PNH, their application in aHUS and secondary CM-TMA remains under investigation. Current studies provide hope for improved outcomes and the development of alternative treatment strategies for these patient populations.
Acknowledgement
J.J.S. is supported by the National Heart, Lung, and Blood Institute/National Institutes of Health (R01HL151367).
Authorship
Contribution: T.P.K. wrote the body of the manuscript and discussion and designed the figures; H.K. wrote the abstract and introduction; K.M. wrote the body of the manuscript and discussion; T.G.D. contributed to the discussion; J.J.S. contributed to the body of the manuscript and discussion; and A.M. contributed to the visual abstract.
Conflict-of-interest disclosure: J.J.S. reports receiving consulting fees from Aronora Inc. The remaining authors declare no competing financial interests.
Correspondence: Thalia Padilla Kelley, Department of Internal Medicine, Oregon Health & Science University, 3181 SW Sam Jackson Park Rd, Portland, OR 97239; email: padillat@ohsu.edu.
